Executing a BLAST Workflow Template
The instructions on this page will show you how to execute a BLAST query workflow using the workflow catalog with the Fuzzball web UI and CLI.
Please select either the web UI or CLI tab to see the appropriate instructions for your environment.
You can run the BLAST workflow template with your data or example data from the workflow catalog page by locating the BLAST card with the CIQ badge and clicking "Run"
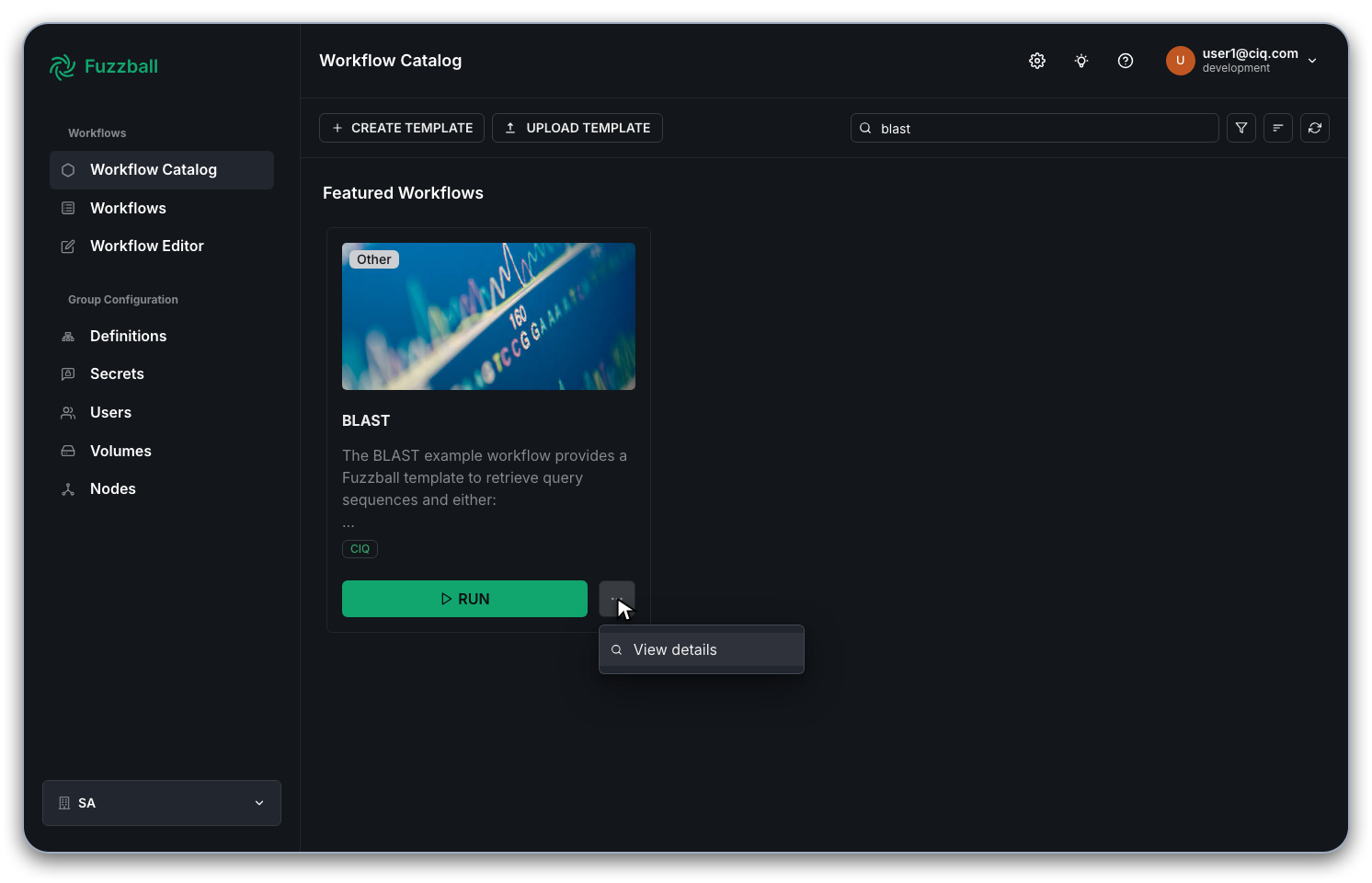
You will be prompted to supply values for the configurable options of the template. You can examine all the options and their documentation. For example:
- The workflow template uses an ephemeral
ScratchVolumeand a persistentDataVolume. - You can use a public database by providing a
BlastDbNamethat matches one of the databases available from the NCBI (see the NCBI metadata json file for all available public options). Alternatively, if you previously created a custom BLAST database and saved it toBlastDbPathon the persistent volume you can also refer to that database here. - If you need to create a custom database you can use the
CustomBlastDbFetchCmdwhich is mutually exclusive withBlastDbName. You can provide any command that outputs a fasta format sequence file that will be used to create a custom database. This could be, for example, anefetchcommand that downloads a number of sequences from NCBI. - The
BlastCmdto run (e.g.blastpfor protein queries/databases) - Any
BlastOptsother than the automatically set input/output/threads - Resources (cores, memory, runtime)
- The path, relative to the root of
DataVolumewhere data should be saved.
If you keep all the default options you will run a BLAST query of some proteins sequences against
the public pdbaa database which includes all protein sequences from the Protein Data
Bank. Click "Run" to render and validate the workflow template with your options.
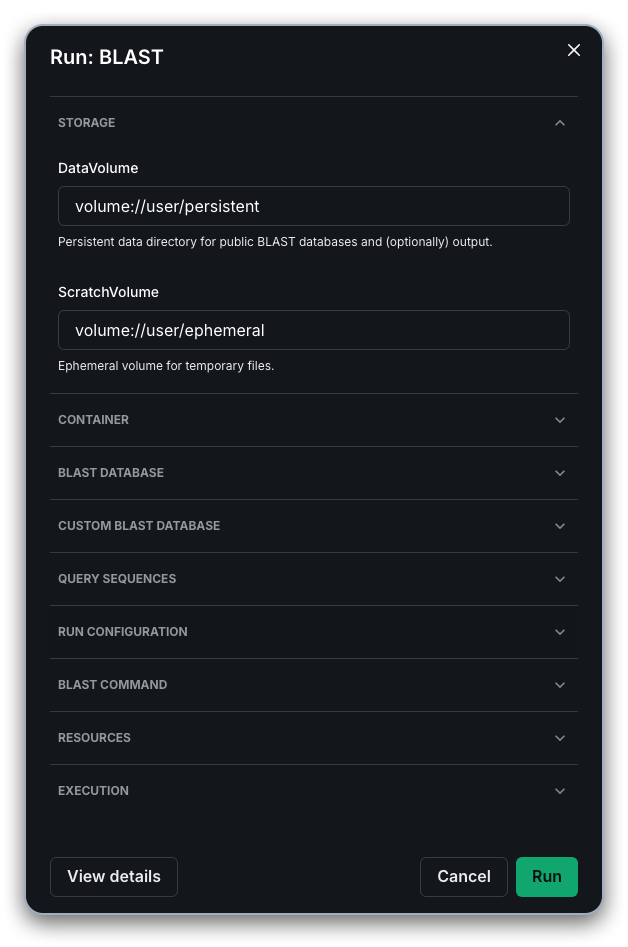
After clicking "Run", you will be prompted in a dialog box to "Start Workflow" to submit the Fuzzfile rendered from the workflow template and your inputs. At this stage you can modify the name of the run or accept the default.
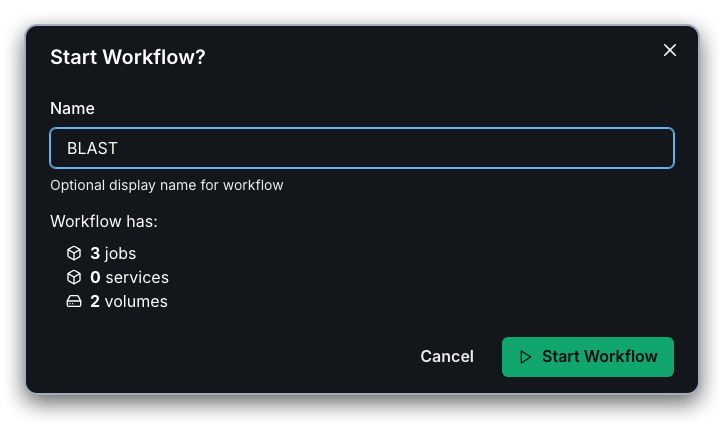
Once the workflow has been submitted successfully, you can select "Go to status" in the next dialog to view the status of the workflow's stages
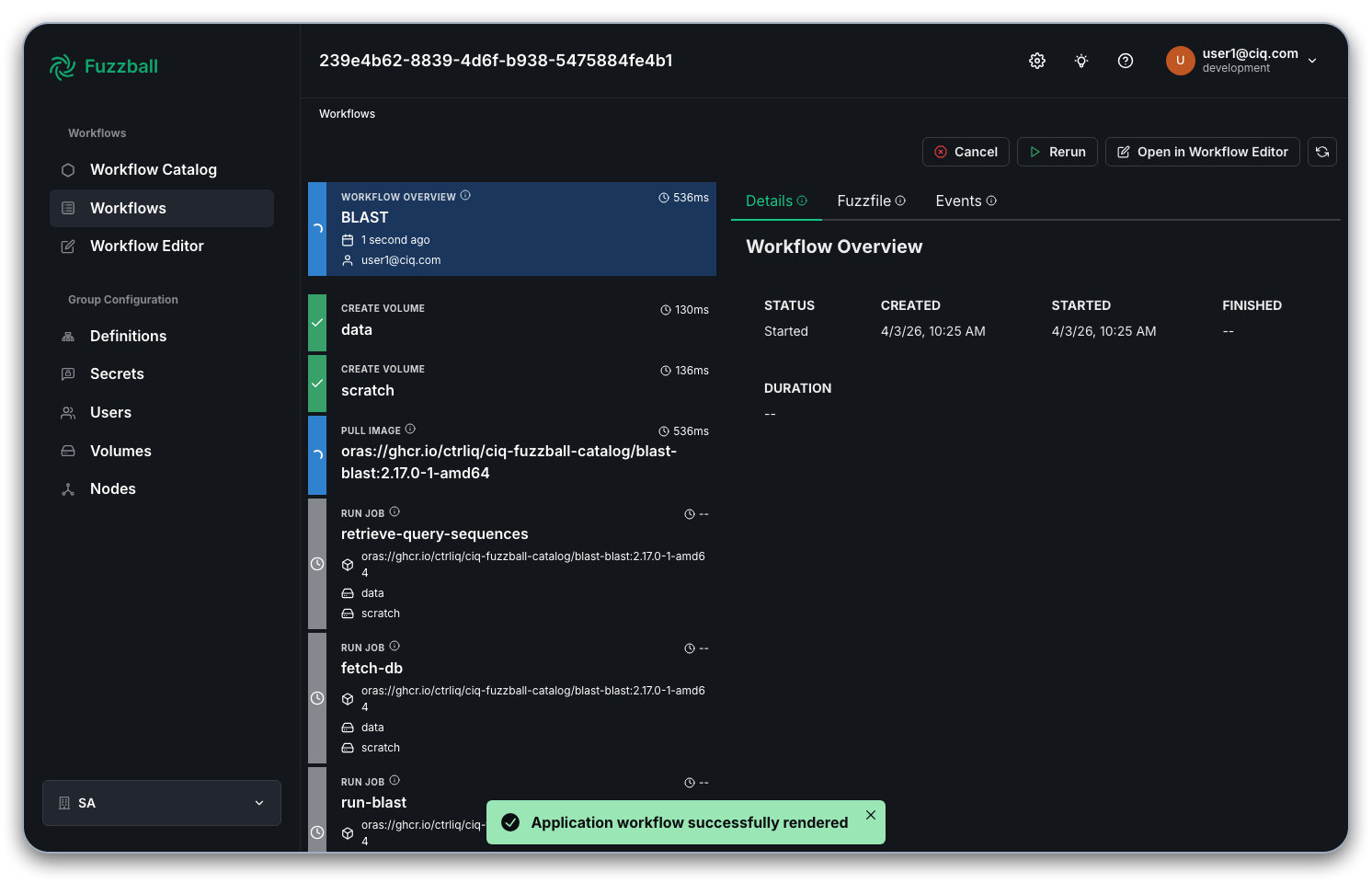
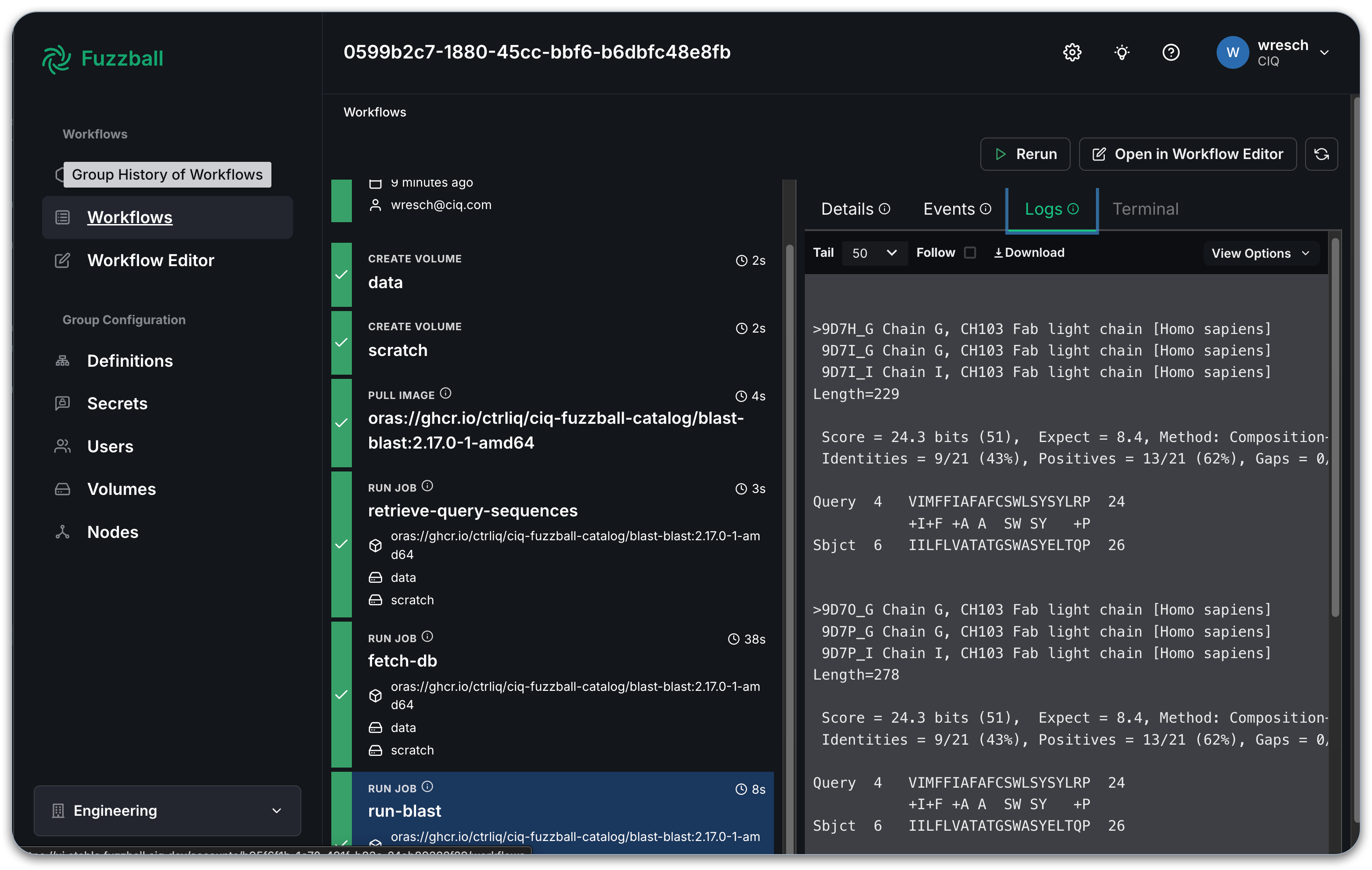
Select a stage that produces output and choose the "Logs" tab to see the output generated by the
stage as we did in an
earlier example
with a manually written workflow. In the example below, we see the beginning of BLAST output from the
run-blast stage
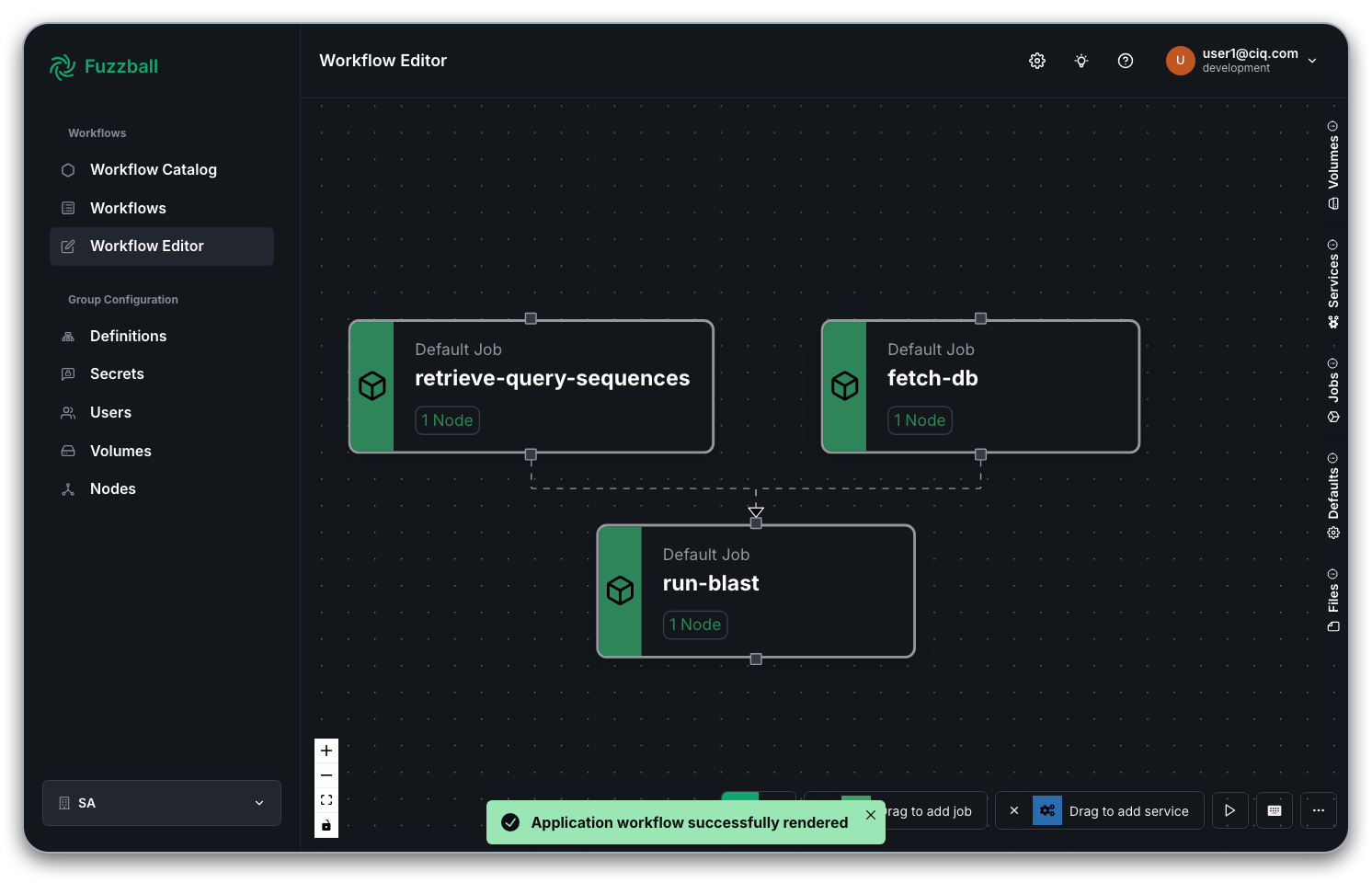
By clicking on "Open in Workflow Editor" in the top right corner, you can open the Fuzzfile generated from the template and your inputs in the workflow editor. In the example above with a public BLAST database the graph of jobs looks like so:
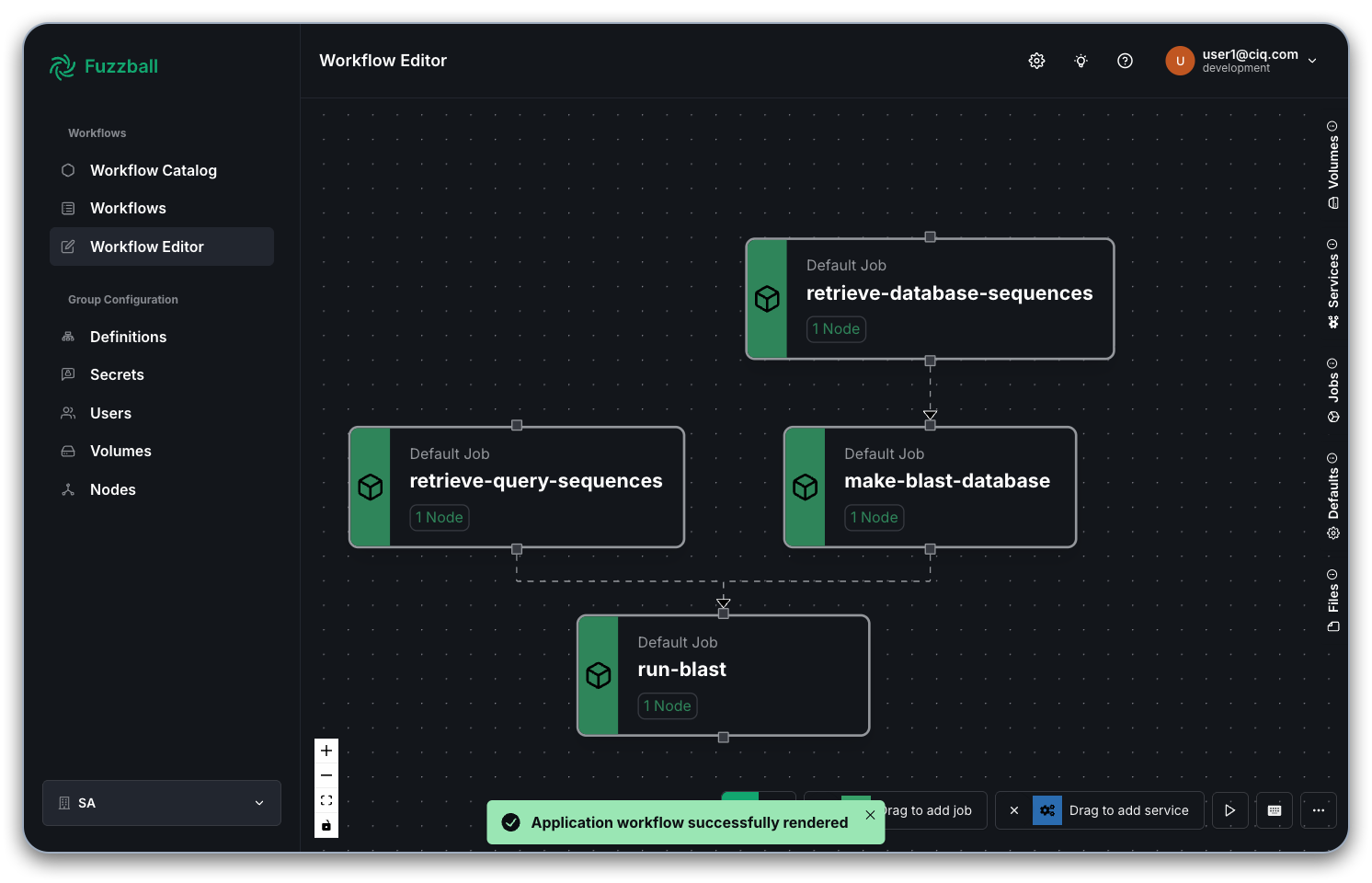
If, instead, you had chosen to create a custom BLAST database the Fuzzfile would have replaced the single job to download/update a public BLAST database with 2 jobs to fetch and build a custom database instead.
To run this workflow through the CLI you will need access to the Fuzzball CLI. You can install it using the Fuzzball CLI installation instructions.
When using the CLI to execute workflow templates from the workflow catalog you need to supply parameters in the form of a yaml file. For the BLAST workflow querying some protein sequences against a public database you can create this file like so:
$ cat > values.yaml <<EOF
values:
- name: "DataVolumeName"
string_value: "testuser"
- name: "DataVolumeProvisioner"
string_value: "persistent"
- name: "ScratchVolumeProvisioner"
string_value: "ephemeral"
- name: "WorkflowContainer"
string_value: "docker://community.wave.seqera.io/library/blast_entrez-direct:2443e1cf34bc04d8"
- name: "BlastDbName"
string_value: "pdbaa"
- name: "BlastDbPath"
string_value: "refdb/blast"
- name: "BlastFetchTimeout"
string_value: "4h"
- name: "RetrieveQuerySequencesCmd"
string_value: "efetch -db protein -format fasta -id YP_232930.1,YP_232961.1,YP_232969.1,YP_232982.1,YP_232983.1,YP_232915.1,YP_232916.1,YP_232979.1,YP_232970.1,YP_232974.1,YP_910498.1"
- name: "RunName"
string_value: "pox_efc"
- name: "BlastOutputPath"
string_value: "results/blast/${FB_WORKFLOW_ID}"
- name: "BlastCmd"
string_value: "blastp"
- name: "BlastOpts"
string_value: ""
- name: "BlastCores"
uint_value: 8
- name: "BlastMemory"
string_value: "30GiB"
- name: "BlastQueryTimeout"
string_value: "4h"
EOF
Note that we skipped some unnecessary parameters that would be used for a custom BLAST database build. Next you need to obtain the ID of the BLAST workflow template. That can be done in a few different ways. For example:
$ fuzzball workflow catalog list
ID | NAME
1767f241-c9ad-44ae-a2b6-e1edbf00770d SomeApp
...
00000001-aaaa-bbbb-cccc-dddddddddddd Hello World (example)
00000002-aaaa-bbbb-cccc-dddddddddddd Jupyter Notebook
00000003-aaaa-bbbb-cccc-dddddddddddd Jupyter Notebook (VDI)
00000004-aaaa-bbbb-cccc-dddddddddddd ParaView
00000005-aaaa-bbbb-cccc-dddddddddddd Xfce Desktop Environment
00000006-aaaa-bbbb-cccc-dddddddddddd LAMMPS (CPU)
00000007-aaaa-bbbb-cccc-dddddddddddd LAMMPS (GPU)
00000008-aaaa-bbbb-cccc-dddddddddddd BLAST
00000009-aaaa-bbbb-cccc-dddddddddddd Stable Diffusion Text to Image
$ id="$(fuzzball workflow catalog list --provider CIQ --name BLAST | awk -F'|' '$2 ~ /BLAST/{print $1}' | tr -d ' ')"
Or if you have jq installed you could make use of the option to return json format metadata about
all templates as shown below:
$ id="$(fuzzball workflow catalog list -o json | jq -r '.[] | select(.name == "BLAST" and .provider=="CIQ") | .id')"
Or you can copy and paste the workflow template id instead of assigning it to a variable. Once you have the id of the workflow template and a values file you can use them to create a Fuzzfile for submission like so:
$ fuzzball workflow catalog render --from-file $id --values values.yaml | awk '/^version/{p=1} p==1' > blast.fz
$ head blast.fz
version: v4
volumes:
data:
use: persistent
name: testuser
scratch:
use: ephemeral
size: 1GB
jobs:
fetch-db:
image:
uri: docker://community.wave.seqera.io/library/blast_entrez-direct:2443e1cf34bc04d8
Note that we used awk to remove any content above the initial version line in case there were any
lines at the top.
The Fuzzfile is then submitted and monitored as described previously like so:
$ fuzzball workflow start blast.fz
Workflow "64888a33-ac09-4fb6-8db6-20aa35fbddc9" started.
$ sleep 10m # or just wait until the submitted workflow is finished
$ fuzzball workflow describe 64888a33-ac09-4fb6-8db6-20aa35fbddc9
Name: blast.fz
Email: wresch@ciq.com
UserId: 87145648-b830-4291-ab7e-40880d61334e
Status: STAGE_STATUS_FINISHED
Cluster: fuzzball-aws-stable
Created: 2025-04-22 01:57:53PM
Started: 2025-04-22 01:57:54PM
Finished: 2025-04-22 02:03:55PM
Error:
KIND | STATUS | NAME | DURATION
Workflow | Finished | 64888a33-ac09-4fb6-8db6-20aa35fbddc9 | 6m2s
Volume | Finished | data | 27s
Volume | Finished | scratch | 27s
Image | Finished | docker://community.wave.seqera.io/library/... | 23s
Job | Finished | fetch-db | 15s
Job | Finished | retrieve-query-sequences | 10s
Job | Finished | run-blast | 14s
$ fuzzball workflow log 64888a33-ac09-4fb6-8db6-20aa35fbddc9 run-blast
BLASTP 2.16.0+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: PDB protein database
170,598 sequences; 48,617,182 total letters
Query= YP_232915.1 serine protease inhibitor-like [Vaccinia virus]
Length=369
Score E
Sequences producing significant alignments: (Bits) Value
4KDS_A Chain A, Plasminogen activator inhibitor 1 [Oncorhynchus m... 153 3e-42
1DB2_A Chain A, PLASMINOGEN ACTIVATOR INHIBITOR-1 [Homo sapiens] 149 6e-41
3EOX_A Chain A, Plasminogen activator inhibitor 1 [Homo sapiens] 149 8e-41
...